98%满意度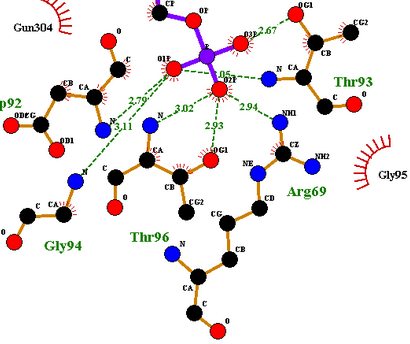
分子对接
平均10个工作日已预约1884次
分子对接
分子对接(molecular docking)是依据配体与受体作用的“锁-钥原理” (lock&key principle),模拟配体小分子与受体生物大分子相互作用的一种技术方法。
配体与受体相互作用是分子识别的过程,主要包括静电作用、氢键作用、疏水作用、范德华作用等。通过计算,可以预测两者间的结合模式和亲和力。

分子对接方法的两大课题是分子之间的空间识别和能量识别。空间匹配是分子间发生相互作用的基础,能量匹配是分子间保持稳定结合的基础。对于几何匹配的计算,通常采用格点计算、片断生长等方法,能量计算则使用模拟退火、遗传算法等方法。根据简化的程度和方式,可以将分子对接方法分为三类。其中,刚性对接方法在计算过程中,参与对接的分子构像不发生变化,仅改变分子的空间位置与姿态,其简化程度最高,计算量相对较小,适合于处理大分子之间的对接。

分子对接方法不仅在药物设计和发现领域得到了广泛应用,还在酶发现与催化领域发挥了重要作用。例如,通过分子对接,可以预测小分子化合物与酶之间的结合模式和亲和力,从而筛选出具有潜在催化活性的化合物,为酶底物的发现提供理论指导。
需要撰写需求文件,说明分子对接需求,并提前与工程师沟通联系。
一、如果计算结果与预期不符合怎么办?
1.科研计算服务是一个探索的过程,其中存在不确定性。服务方对理论计算得到的数据负责, 但不保证理论数据能够与预期结果完全一致。如果需要可以对模型进行适当调整,调整过程中若增加计算或者大规模的重新计算,需增加计算费用,价格另行协商。
2.模拟计算存在不确定性,我们对计算过程的正确性负责。计算开始之后会消耗软件,机时以及人力成本,所以不支持退款,敬请谅解
二、模拟计算项目服务范围?
1.计算完成之后提供所需的计算方法,计算结果数据和图文,以及关于计算结果中疑问的解答;
2.文章回复意见中模拟计算问题进行适当的解答,若返稿后需要增加计算,需另行收费。
三、关于模拟计算下单流程是怎么样的呢?
1.如果是确定计算需求,可以直接在测试狗官网选择相应的项目进行下单,提交订单时需填写具体的计算内容,预期结构,分子结构及相关的参考文献。如果网站上没有对应的项目请选择【私人定制】并在需求中写明具体计算内容;
2.如果不能确定计算内容,可以直接联系工作人员进行需求沟通。